This preproof from Nathan Grubaugh (Yale, PhD in microbiology, 2016), Bill Hanage (Harvard, PhD in epidemiology), and Angela Rasmussen (Columbia, PhD in microbiology, 2009) came across my desk. It’s about the spike protein variant that, as Bette Korber at Los Alamos demonstrated, has become the dominant strain of the coronavirus in the last couple of months.
These guys present no data, just opinion. Here’s the core bit of their paper:
[Korber et al.] present compelling data that an amino acid change in the virus’ spike protein, D614G, emerged early during the pandemic, and viruses containing G614 are now dominant in many places around the world. The crucial questions are whether this is the result of natural selection, and what it means for the COVID-19 pandemic. Korber et al. hypothesized that the rapid spread of G614 was because it is more infectious than D614…As an alternative hypothesis… the increase in the frequency of G614 may be explained by chance, and the epidemiology of the pandemic.
In February, the area with the most COVID-19 cases shifted from China to Europe, and then in March on to the US. As this and other work shows, the great majority of SARS-CoV-2 lineages in the US arrived from Europe, which is unsurprising considering the amounts of travel between the continents. Whether lineages become established in a region is a function not only of transmission, but also the number of times they are introduced. There is good evidence that for SARS-CoV-2, a minority of infections are responsible for the majority of transmission (Endo et al., 2020). Therefore, while most introductions go extinct, those that make it, make it big (Lloyd-Smith et al., 2005). Over the period that G614 became the global majority variant, the number of introductions from China where D614 was still dominant were declining, while those from Europe climbed. This alone might explain the apparent success of G614.
Got that? They think that random chance is a likely hypothesis. Europe happened to be the next big outbreak after China, Europe happened to get this G614 variant, and then Europe spread that variant to the rest of the world.
There are some historical problems with this account. But before I get to those, I want to go through some math. How likely is it that a new introduction of the virus “succeeds” – that is, seeds an infection chain that doesn’t die out? We can approximate this based on three parameters:
- The relative advantage
of the introduced strain as compared with the old strain.
- The growth rate in the new location
, which gives the mean new infections per infection.
- The overdispersion
, which describes the variance in new infections per infection.
The assumptions I’ll need for the approximation are that
Novel Introduction
First, let’s estimate how likely it is that a single infection can seed a completely new outbreak in a new location – one where there isn’t another outbreak already. I won’t go through the details here, but we get

We can then use Haldane’s approximation to estimate how many introductions are needed to get a 50% chance of seeding a new outbreak:

A couple of points: if the mean growth rate in the new location is less than 1, no new outbreaks. You could have an unlucky first “superspreader” event, but then those secondary infections would on average die out. The higher the growth rate is, the more likely the outbreak is to seed. Second, the overdispersion makes the infection harder to seed relative to a Poisson distribution. The more variable the growth rate is, the less likely any single infection is to seed a big event.
To feed in some actual numbers, if 
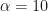


Introduction Under Competition
What happens if we try to seed an outbreak where there’s already cases? Depends on the difference in growth rate between the existing cases and the new strain. Here 

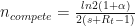
Increased fitness makes it more likely. Note that if the local growth rate is negative, you can still get a new outbreak if the higher fitness would push the growth rate positive.
History
Alright, let’s talk West Coast numbers. I’ll be using nextstrain.org, since that data’s publicly available. The West Coast (California, Oregon, and Washington) had very early outbreaks, seeded by travelers from China with the original D614 strain. By May 14, there are no more sequenced genomes of the original D614 in the database. The last one in Washington is May 6, in California May 12, and in Oregon May 13. Back in February, the original strain was the only one sequenced in Washington, and the dominant one in both Oregon and California. Note that Washington then was the center of the US epidemic, with several deaths by the end of February. The first of the new strain to show up in Washington is March 11, Oregon March 11, and California February 4. If we focus on Washington, that means in two months (or about 10 generations, with a 6 day generation time) the new strain completely takes over.
Now, the test volume in Washington was about 4000-5000 tests per weekday over the whole of that two months, with about 200-450 positive tests per day, peaking in late March and then dropping slowly until early May. That suggests the growth rate was slightly negative during that two months, making it very hard indeed to seed a new outbreak of the new strain from only a couple introductions.
Strain Replacement
Just introduction of the new strain by New Yorkers or Europeans won’t cause complete replacement of the old strain by itself. If there’s no fitness differential, all it will do is seed new infection chains with the new strain. Suppose you have 500 cases of the old strain and then 4,500 New Yorkers come to Seattle. Then most of the new cases will be of the new strain, but not all of them. There’s no reason for the old cases to stop presenting new cases.
One way for this to happen is if there is a differential fitness, so that the old strain has a negative growth rate while the new strain has a positive or less negative growth rate. That lets the new strain outcompete the old strain, until the population of the old strain is small enough that it can disappear by chance.
The other way is for all the existing cases to get swamped by new introductions under conditions of very low growth. Suppose the intrinsic growth rate in Washington was extremely low, say 0.5. So the number of cases drops by half every generation. But every generation new cases get introduced from another location dominated by the new strain (e.g., New York). So we start off with say, 400 cases in Washington. Then in the next generation, those cases only generate 200 new cases. But we also get another 150 cases imported from New York, for 350 total cases, 43% new. Repeat for 10 generations, and you’ll end up with no cases of the old strain, but a fair number of cases of the new. Note that you need a really low growth rate for this to happen in only 10 generations: 
