Introduction
What happens to a new mutation?
Every person has some new mutations in their somatic genome, mostly caused by errors in DNA replication. They can pass that mutation on to their descendants. We know of a couple of historical mutations that look like they are all from a single common ancestor – for example, the common variant that’s responsible for blue eyes. How likely is a new mutation to succeed and become common?
This depends on the selective advantage 


We’re most interested in humans right now, and humans are diploid; they have two copies of each chromosome. Any variant can either be on both chromosomes or only one, and the selective advantage can be different depending on this. There are many common variants that are helpful when only one copy is present and harmful when two copies are present. Whether a mutation succeeds and becomes common mostly depends on the selective advantage in carriers with a single copy, because carriers with two copies are very rare when the mutation is uncommon.
There have been multiple approaches to solving this problem. Today we’ll look at one of the earliest and most simple. It’s straightforward, but requires a couple of assumptions that are not always valid. But we can get a lot of utility out of the simple result.
Haldane’s approach
Haldane’s original 1927 paper has a derivation of the likelihood of a new mutation becoming common, but it can be somewhat difficult to understand. We’ll work out his results in slightly more detail.
Suppose we have a large population with a new mutation present in a single individual. Let’s let 


We’ll see why this function is useful in more detail later. By construction, the coefficient of 


![[f(x)]^m](https://s0.wp.com/latex.php?latex=%5Bf%28x%29%5D%5Em&bg=ffffff&fg=111111&s=0&c=20201002)
Expanding on this a bit, suppose that 


If we have two carriers,
![[f(x)]^m = 0.25 + 0.5x + 0.25x^2](https://s0.wp.com/latex.php?latex=%5Bf%28x%29%5D%5Em+%3D+0.25+%2B+0.5x+%2B+0.25x%5E2&bg=ffffff&fg=111111&s=0&c=20201002)
And we can see that the coefficients of the powers of x compose the probability distribution of carriers in the next generation.
Now, what would happen if instead of multiplying the function, we iteratively apply it to itself? This forms a composite function

Here the superscript 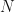

We can see that this function 
We can then represent the likelihood that the variant disappears from the population as 

This kind of repeated application of a function to an input is called a fixed point, and it will converge to a root of the equation 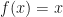
.
Now, to be able to go further, we have to make some assumptions about the form of the function 

This can be simplified with some algebraic work I won’t show here to

So the probability of extinction is 
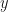

So let’s plug 


We take the natural log of both sides to get

Then we can take the Taylor series of 

Or simplify to give

Now, if the selective advantage 
Implications
First let’s remember the assumptions that went into this calculation:
- The population size is “big”, so that we can ignore the likelihood of carrier-carrier matings.
- The number of surviving children per individual is Poisson-distributed.
The biggest implication of this result is that the likelihood of the mutation surviving doesn’t depend on the size of the population! It’s just as easy for a mutation to become common in a small population as a large population.
Second, the likelihood of survival is directly proportional to the selective advantage of the mutation. But since even highly adaptive mutations have low absolute values of
Finally, if you repeatedly introduce the same beneficial mutation into a population, it’s very likely to become common. This suggests that even small amounts of crossbreeding will result in beneficial mutations from one population becoming common in the other population.
